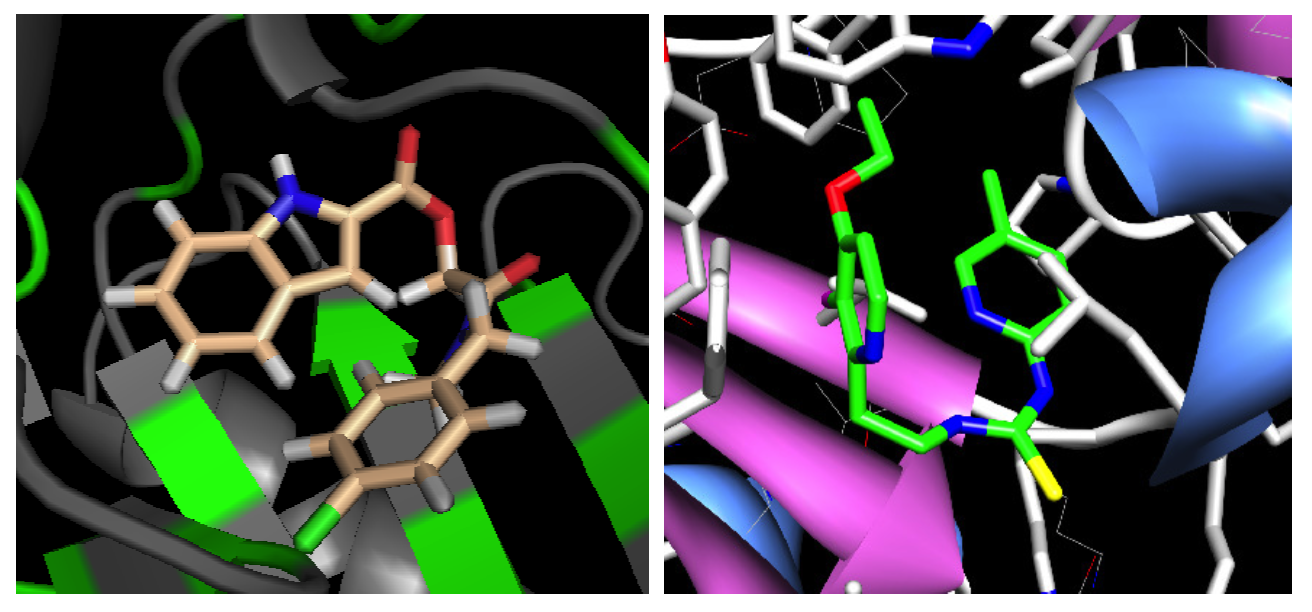
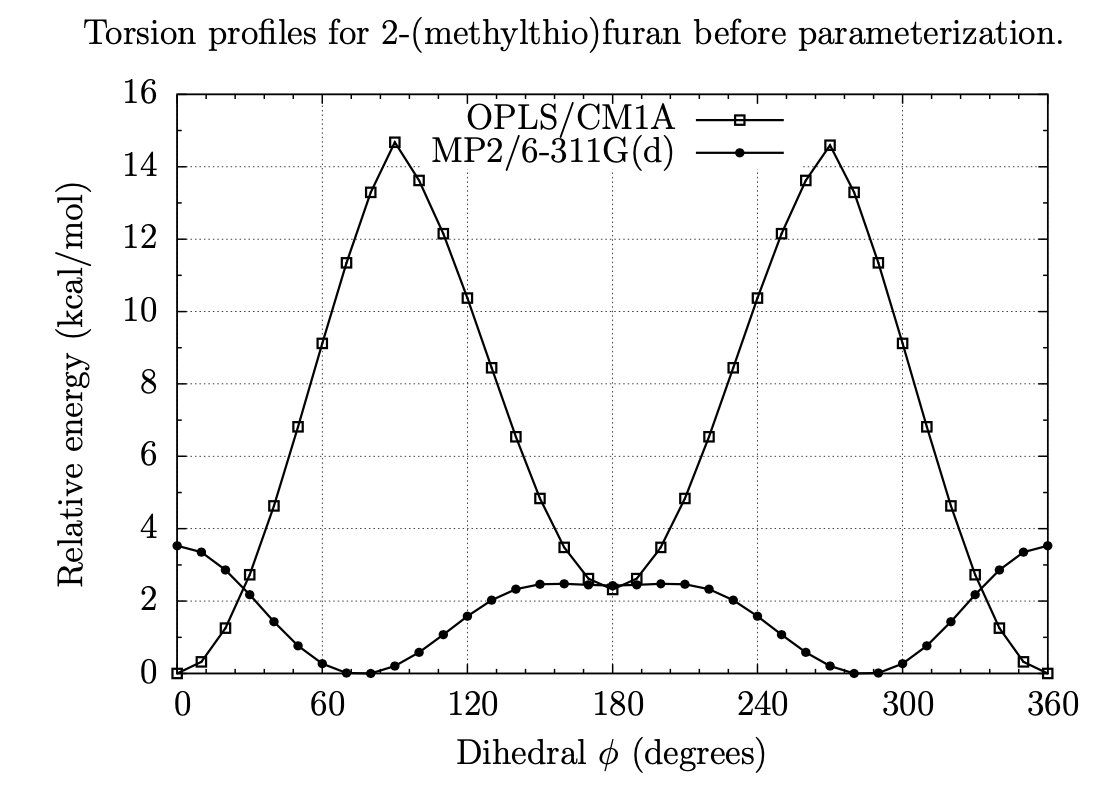
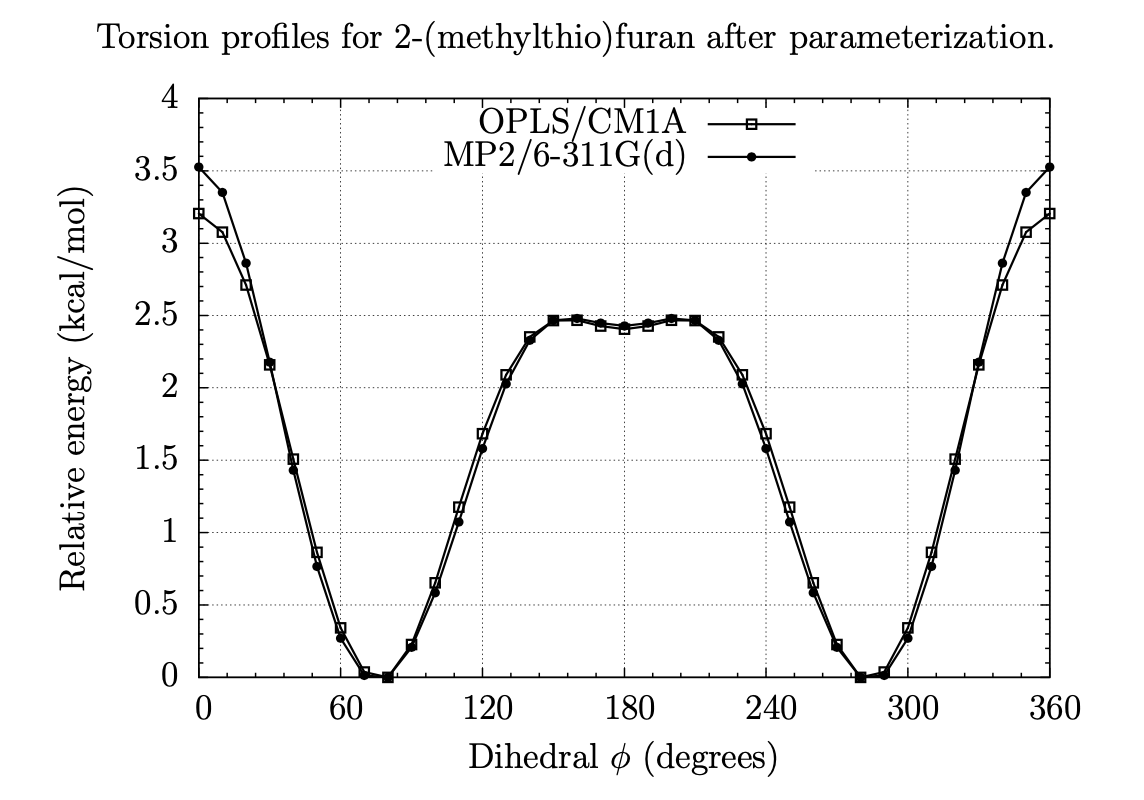
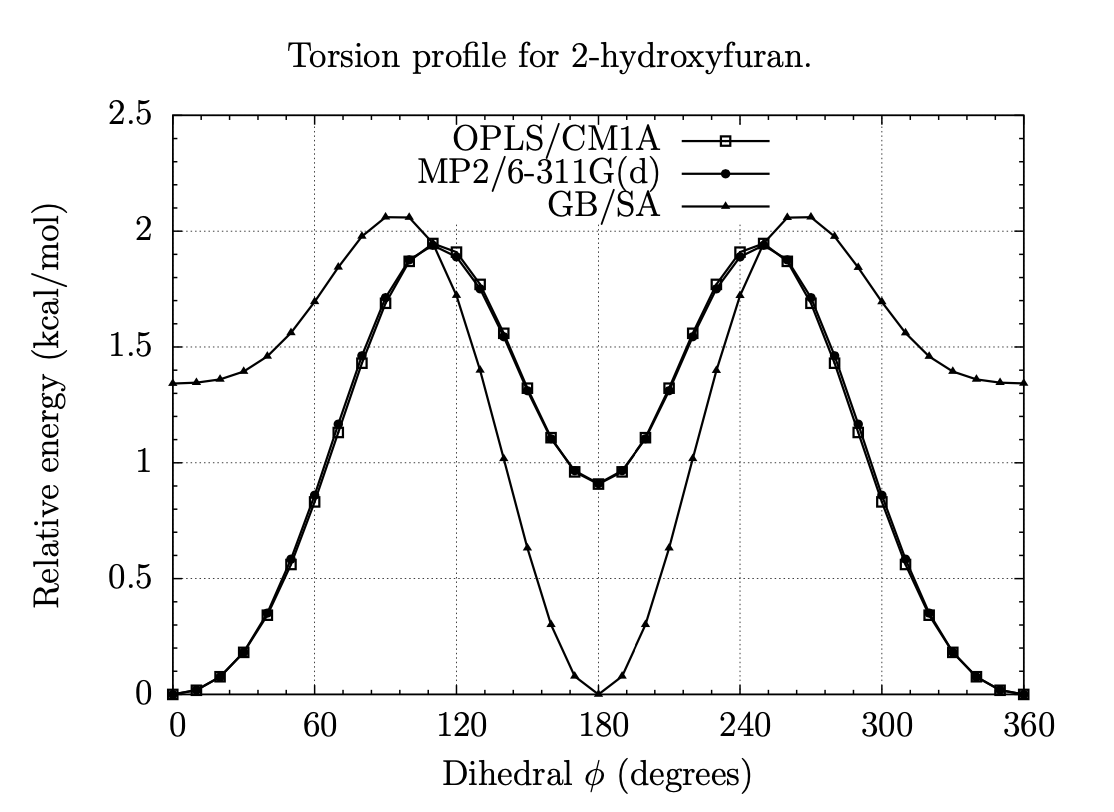
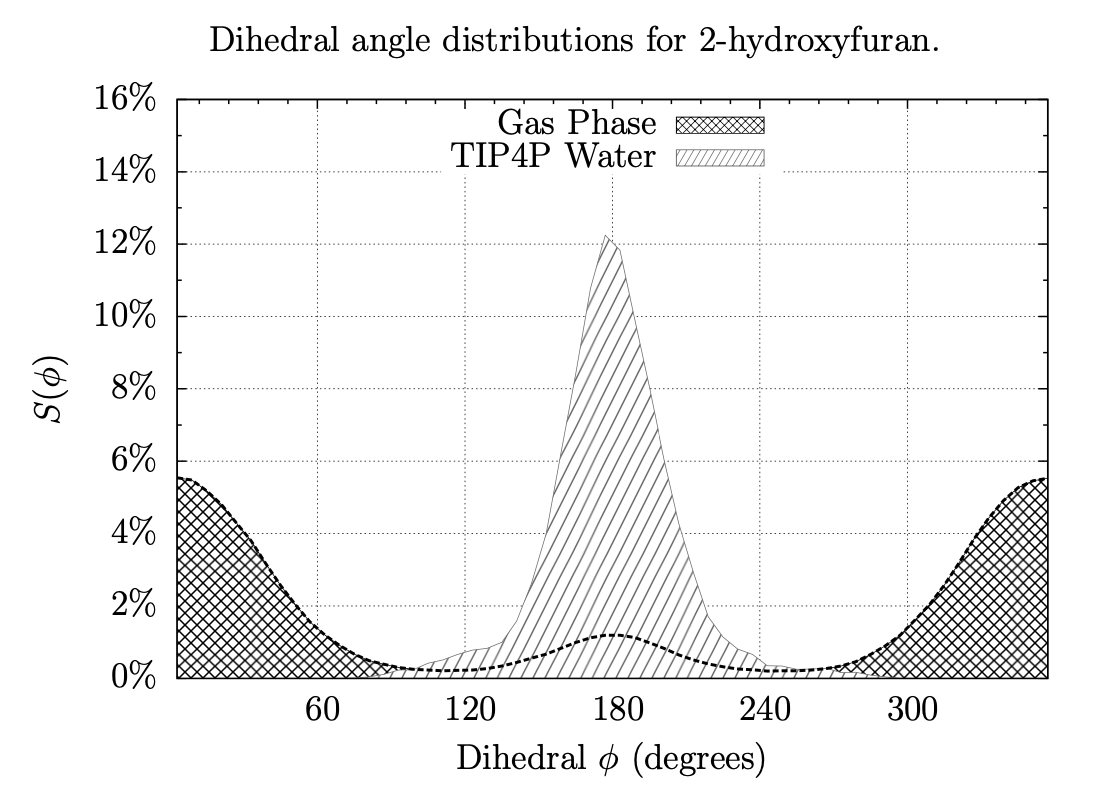
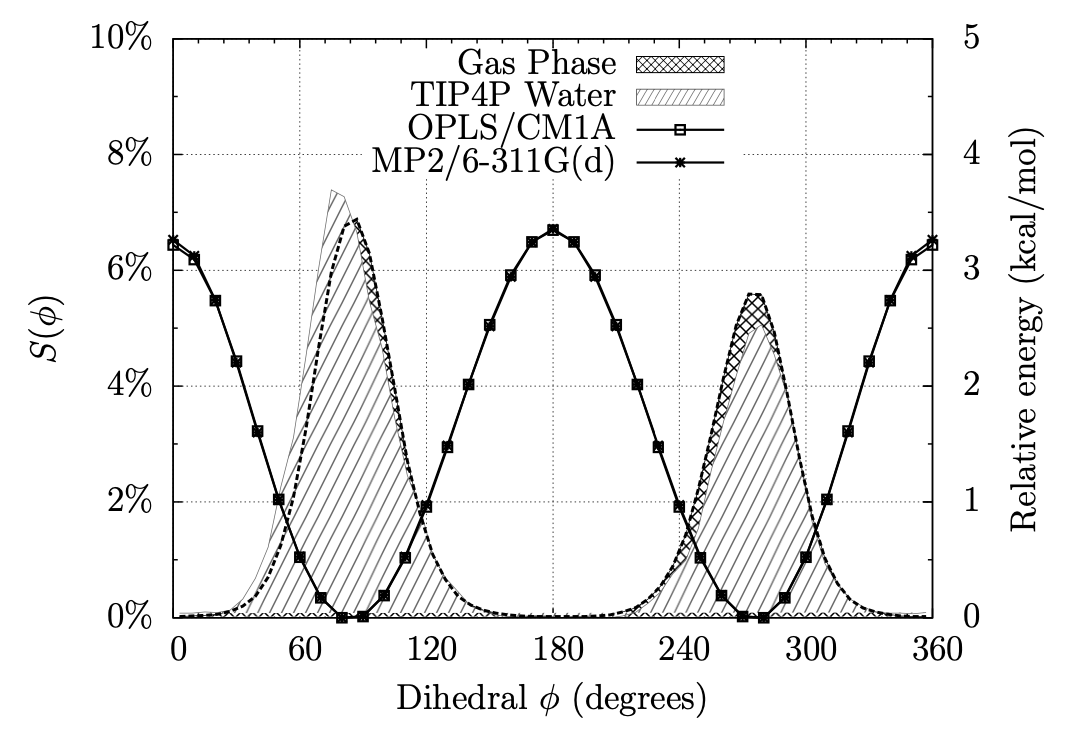
A Fragment-Based Search for HIV Inhibitors using Linear-Response Property Prediction
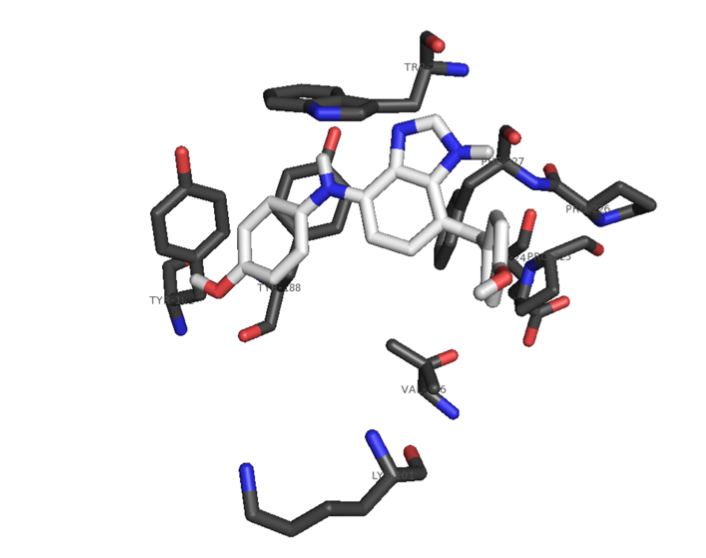
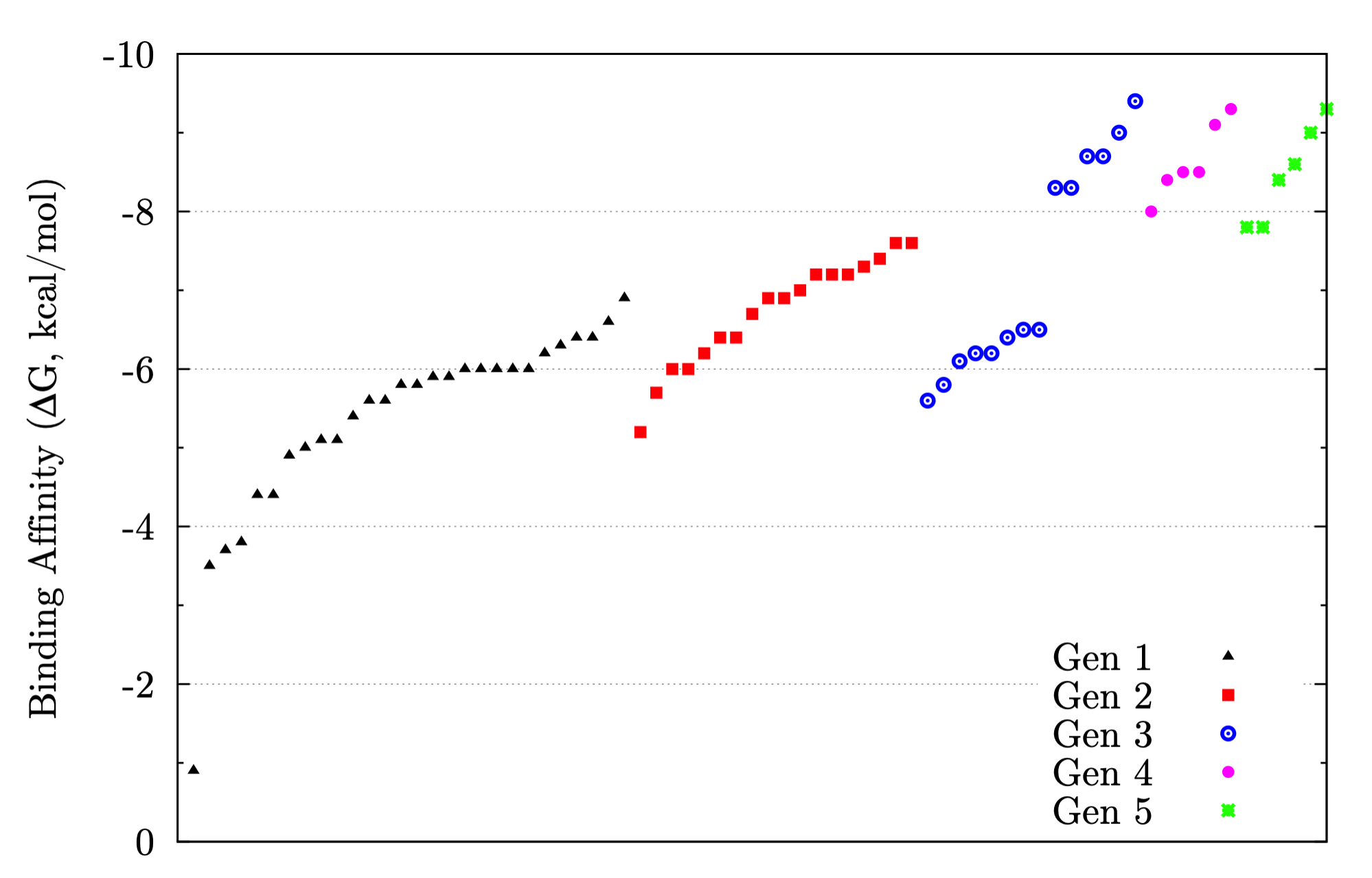
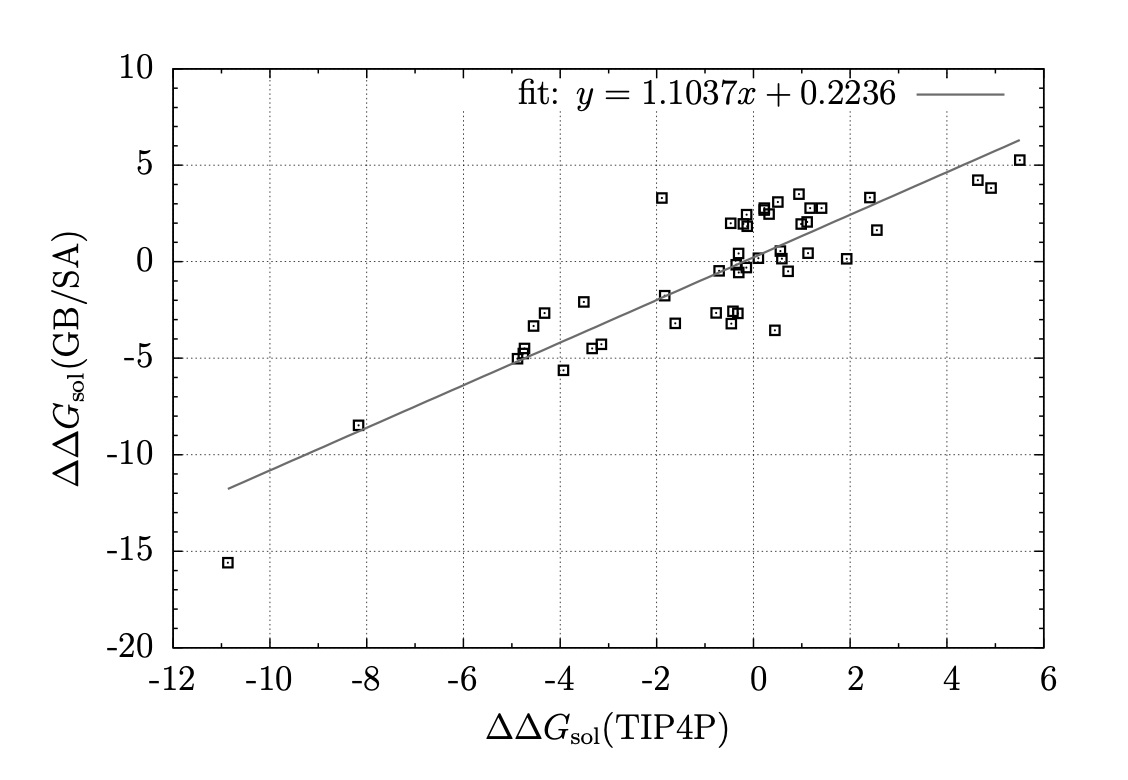
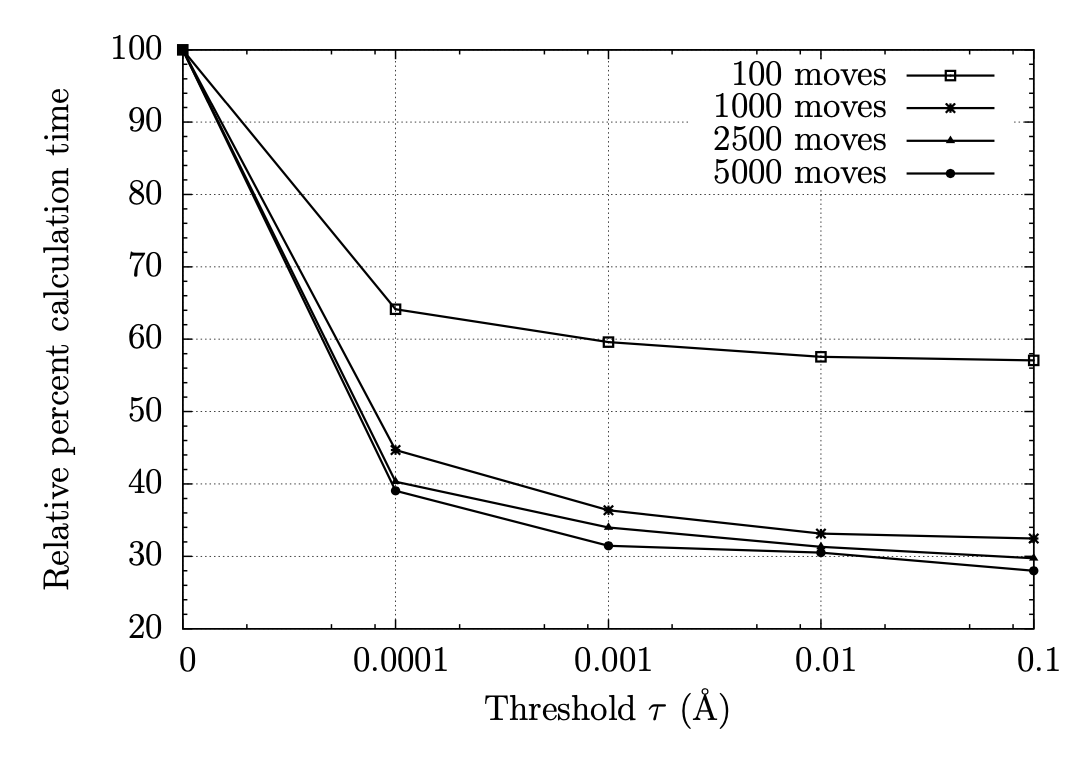
The foundation of any QSAR analysis begins with the fragment library: a database of molecular fragments and their associated geometric, physical, and chemical properties. This project involves design and population of a fragment library for discovery of inhibitors of HIV-1 reverse transcriptase, a key enzyme in the retroviral reproductive pathway. Research articles published in 2011 and 2012 indicate that heterocyclic cores connected by various linker groups can bind irreversibly to an allosteric site on HIV-RT, but novel structures such as these have not been widely studied in a systematic fashion. Our goal is to design a large set of heterocycles, linker groups, and derivatives, then to characterize them as individual fragments and as combinations thereof. Once the library is complete, we will use it to design drug candidates that should bind to our target based on complementary geometries, torsions, polarities, and hydrogen bonding; docking our candidates into the allosteric binding pocket of HIV-RT will then allow us to rank our results quantitatively.